Overview
During a recent panel discussion, members of the Flagship Biosciences team and representatives from The Johns Hopkins University, Leap Therapeutics, and Corritori Consulting shared their thoughts on companion diagnostics development, with an emphasis on oncology. They spoke about what the regulatory approval pathways entail, as well as points to consider for tissue-based companion diagnostics.
The panel also discussed image analysis, performance criteria, and additional regulatory considerations that anyone developing a therapeutic may want to take into consideration, particularly as you move forward into trying to gain acceptance by the U.S. Food and Drug Administration (FDA).
 Special Thanks to Our Panelists:
Special Thanks to Our Panelists:
- Susana Corritori, Regulatory Consultant, Corritori Consulting
- Roberto Gianani, Chief Medical Officer, Flagship Biosciences
- Mike Kagey, Senior Director of Translational Medicine, Leap Therapeutics
- David Sidransky, Professor at The Johns Hopkins University School of Medicine
- Meredith James, Chief Operating Officer, Flagship Biosciences (moderator)
Introduction
Companion diagnostics are used to support the safe and effective use of a corresponding pharmaceutical product. A companion diagnostic is an in vitro diagnostic test device that has the information that is essential for the safe and effective use of the therapeutic product. It helps to identify suitable patients who are eligible for a particular targeted treatment and supports monitoring of the state of response.
The stipulated instructions for use of both the companion diagnostic and the corresponding pure diagnostic are their safe and effective use. Ideally, a therapeutic product and corresponding companion diagnostic are co-developed. They may start at the same time and may be determined by the speed of drug development.
 Often contemporaneous approval may not be possible, which is recognized by the FDA. A common situation is that the approval of a therapeutic product occurs first and then the approval of a companion diagnostic takes place, requiring potentially in addition, the labeling of the therapeutic product in the post-approval process.
Often contemporaneous approval may not be possible, which is recognized by the FDA. A common situation is that the approval of a therapeutic product occurs first and then the approval of a companion diagnostic takes place, requiring potentially in addition, the labeling of the therapeutic product in the post-approval process.
Another is that a new marketing indication is possible for an approved drug with a new diagnostic. Regulatory oversight, in any case, is risk-based.
It is prudent to start investing and evaluating the companion diagnostic pathway as early as possible. It is a lengthy process, going from a hypothesis all the way through CDx development. The earlier that you can begin to consider the process ensures that there is more time to react to the inevitable issues that come up and allow you to plan for the future.
Your biomarker companion diagnostic development should also be closely linked to your drug development.
You may also view our recorded presentation about these topics. We are happy to provide a consultation on your project. If you need assistance in planning your sample and regulatory approaches and strategy, Flagship can help, too. Contact us to learn more.
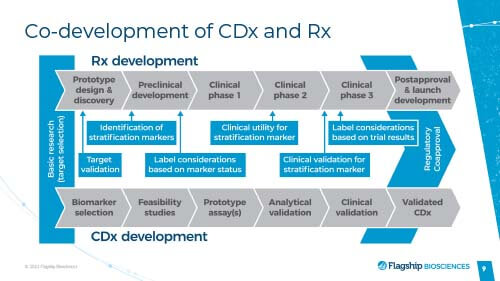
Co-development of CDx and Rx
If you are developing a drug based on a biomarker strategy, where eventually you are going to stratify patients or use the biomarker to select or identify patients, your assay will eventually need to be FDA approved, too.
One example, at a high level, is the process that Leap Therapeutics took for the companion diagnostic that they are currently developing. One of their lead oncology assets is a therapeutic antibody called DKK1 that targets a secreted protein, DKN-01.
They divided their CDx development into multiple stages, including:
– A non-clinical, hypothesis-generating stage, where they came up with models and hypotheses about what potential biomarkers could be used for their drug.
- In their case, the most obvious was the target of the drug, the protein DKK1, that drove tumor growth. They hypothesized that patients who are producing more of this protein in the tumors were the ones who would be able to respond.
– The next step was to determine what assay they would use to measure DKK1 levels.
- They tried IHC for protein but couldn’t get that to work
- They then moved on to an RNAscope® asset to measure DKK1 mRNA levels
- At this point, they wanted an assay that would be reliable, but it did not need to be validated or verified extensively
- To get to a laboratory-developed test (LDT) is much more of a financial commitment
- For them, as they were testing a variety of different hypotheses, they just needed a solid assay, basically a research use only (RUO) assay.
- They did some internal validation and got the assay up and running.
– The next step was moving into testing some clinical samples
- They did a retrospective analysis from tumor samples that they had already collected during Phase One trials, trying to parallel the clinical development of their drug.
- They took the RNAscope assay, ran it on the clinical samples, and were able to show that patients who were expressing levels of the target of the drug were the patients who were deriving the most clinical benefits.
- After seeing this data, they were at an inflection point, with data that they thought was strong enough to move the assay to the next step of the companion diagnostic pipeline.
– For Leap, their next step was using the assay to prospectively screen for patients.
- It required the assistance of a partner, which is when they started working with Flagship Biosciences
- They needed their assay to be validated and demonstrated to be reproducible, accurate, sensitive, and specific assay, all validated to CLIA and CAP guidelines
- It also needed to be run in a single lab, to be used prospectively to identify patients for their clinical trial.
Developing an assay and getting it to a laboratory-developed test level takes time. Paralleling drug development, the assay needed to be up and running for Leap to actually enroll patients. For a small company, it was important to have it running so it didn’t delay the clinical development program.
Leap Therapeutics got the assay up and running and is currently prospectively screening patients for a Phase Two trial.
Though this trial is ongoing, they are planning for success, and they are starting to think more about the next step as they move further along the CDx development.
Their next step is to take the LDT and transition into their registrational in vitro diagnostic.
Think of this as an assay that will be locked down and hopefully, it will be the assay that you will test after the approval of your drug.
For this to happen, additional verification and validation of the assay must occur. You will be using GMP reagents and the assay, in Leap’s case and potentially for most drug development, has to be shown that it can be run in multiple labs to support global, registrational trials.
This is another big jump, from the LDT to the CDx. You are investing a lot more in the assay and want to be sure that you’re ready to do this. At the same time, it takes time to move from that RUO assay to an investigational use only assay that you can use in the registrational Phase Three trials.
Planning is critical. It may further come down to how much you can spend upfront and how aggressive you want to be.
Design and Incorporation of Sample Strategy Support
 From an oncologist’s perspective, the important thing is to make sure that it works, in terms of your needs in general. It is important to try to start at the same time to think about what your CDx is going to look like as you’re developing your therapeutic. It also helps you think about what kind of samples you’re going to need and to be able to act to access them as early as possible.
From an oncologist’s perspective, the important thing is to make sure that it works, in terms of your needs in general. It is important to try to start at the same time to think about what your CDx is going to look like as you’re developing your therapeutic. It also helps you think about what kind of samples you’re going to need and to be able to act to access them as early as possible.
Whether you have blood samples, for example, that you have to take that are not readily available, or fluid samples, or special types of tissue samples that have to be taken, the more you understand what your needs are and when you need them, the better it will be. You need to try to strategize early on as you think about both your therapeutic and diagnostic approach.
The nice thing about tissue-based diagnostics is that, as a matter of proof, almost every patient who has had a biopsy has stored tissue that will be available. To be able to make the diagnosis, you don’t have to maneuver to get an additional kind of sample, which is an advantage of tissue-based diagnostics.
From a development point of view, if you can’t think about it at the same time and get those samples prospectively, that’s OK. It rarely happens that everything works out. Sometimes you have to think about your diagnostic after you have developed a therapeutic. You may have to think through it together with the FDA.
You can sometimes go retrospectively back into samples. Sometimes they have to have an additional trial where you collect the samples and then do the tissue-based diagnostics.
But, again, those are all in discussions with the regulatory agency. The most important takeaway is to do this as early as possible. Your tissue-based diagnostic is going to help you understand how a patient is going to respond to a therapeutic, for example how they are going to take the drug and when they are going to take the drug.
If you think about it later, after the fact, it may be very hard to access the samples and it may be hard to go back in a reasonable amount of time. It will also require a lot less money if you think about and act on this at the beginning of your development.
Talk to the regulatory agency about how your trial is going to look, not so much from the therapeutic point of view, which has already been discussed, but more from how your diagnostic is going to be developed, so you can plan the right strategy.
You may also view our recorded presentation about these topics. We are happy to provide a consultation on your project. If you need assistance in planning your sample and regulatory approaches and strategy, Flagship can help, too. Contact us to learn more.
Regulatory Considerations of Oncology and Companion Diagnostics
It is important to engage in early dialogue with the regulatory agency at a strategic level and then to expand into the implementation of that strategy. In oncology, this is particularly important because of the large number of targeted precision medicines and the risk level associated with companion diagnostics in oncology drugs. Oncology drugs have an inherent risk profile. They are imparting critical life benefits to the patients who take them.
The tissue collection process necessary for laboratory-developed tests and later on, the companion diagnostic, require medically invasive procedures like biopsies. Since the regulation of a companion diagnostic is risk-based, it puts some tissue-based diagnostics, companion diagnostics, into the so-called “Category Three” of risk, which is considered high risk.
A common scenario is that a pre-market application is required in the process of clinical trials to demonstrate required performance, related to the safe and effective use of therapeutic products. For the follow-on approaches for the refinement of a companion diagnostic, you can rely on less stringent regulatory approval pathways such as 510K clearance, where the originally approved CDx serves as a predicate or reference IVD test.
Bridging Your LDT to a Clinical Trial Assay
To continue our earlier example from Leap Therapeutics, they have developed an LDT that is currently being run by Flagship Biosciences to prospectively screen patients that have high tumor suppression of the target of their drug for their Phase Two trial.
As they believe this trial is going to be successful, they are starting to think about how they will transition their assay from an LDT to a registrational clinical trial assay that can be run for a registrational Phase Three trial.
Because of this, they now have to transition from RUO to GMP reagents and it can take time. They are using the RNAscope assay, and they have to work with the developers of the technology to manufacture a lot of the reagent that will be used.
They will eventually want to run their tests in multiple, different labs. They will have a global registrational Phase Three trial and are planning on having at least two different labs.
This moves the assay from an LDT into more of an investigational use only, where you have to demonstrate that the assay is not just reproducible in an accurate and specific sense in one lab, but multiple labs.
With this assay registrational clinical trial agreement (CTA) that Leap wants to run for their pivotal Phase Three trial, they want that assay to be as locked down as possible. They are hoping to start their trial with an assay/test that they will not have to modify for use with the FDA. The idea is to mitigate the need to modify the test halfway through the Phase Two trial.
To demonstrate compatibility, they are saving as many clinical specimens and slides from the current Phase Two trials as possible so that if they must tweak something, they can go back and run the modified assay and demonstrate that the modified assay is performing at a comparable level as the one that they originally ran.
When you do go into a registrational CTA, this also does require another level of verification and validation. It is more rigorous than what you have to do for just an LDT and will require running your biomarker assay on multiple different samples, and these can be samples that you commercially acquire. It can be challenging to find these commercial samples. You want to acquire samples that are going to be very similar, if not identical, to the samples you’d be testing for during your Phase Three trial.
This might be endoscopic or core needle biopsies, but this can be challenging to do. It is important to discuss this with the FDA, to either figure out how to best address what the mitigation can be going forward and what the FDA is comfortable with that. Start thinking about it early, so that you can be as prepared as possible.
For the test to be used for prospective enrollment of patients, it must follow the quality and standards of performance certified under CLIA conditions. This is something that is part of the package that your IRB for the trial will be looking at for assurance that the test, despite the fact of the laboratory nature of development, still satisfies the high level of quality standards that will, later on, further be developed and expanded.
There’s a new framework in the EU for LDTs as well. If you have questions about it, feel free to contact us.
Approval Pathways for New Companion Diagnostic Assays
As previously mentioned, the classification of the companion diagnostics and regulatory oversight is risk-based. Regulatory approval options are also risk-based and depend on whether there is already an approved test on the market, that can serve as a reference test. Or, this is a new drug, a new companion diagnostic, which will have to follow this pathway.
In general, there are three different regulatory pathways for a companion diagnostic, an IVD.
- We mentioned the 510 K pathway, which practically uses existing reference products on the market as equivalent or so-called predicate, in which case, the focus of the regulatory submission is really to establish the equivalency or connection with the predicate. However, no clinical trials usually require this regulatory option.
- Most of the new IVDs and companion diagnostic will follow pre-market approval pathway. This will require a clinical program that will be incorporated into drug approval. It is a complex process, depending on the target, depending on the test, and depending on the disease. It is also the most expensive and requires the most time.
- There is another pathway that is called de novo reclassification when no predicate IVD exists on the market, however, the new IVD or CDX is of low risk or moderate risk, which in most cases, no comprehensive clinical evaluations will be required.
Most companion diagnostics of new drugs will follow the PMA pathway, and for that, early consultations with FDA have to be scheduled to discuss the strategy.
The Clinical Validation Process
So, the clinical validation process is, of course, very important. It can be a complicated and serious undertaking. From the point of view of clinical validation, one important thing is to have a large number of specimens.
Heterogeneity for each indication can be so large that utilizing a large number of specimens will yield a different type of morphology that will create a solution, that can be useful in the real world. That is the analytical reason.
The second reason is that ultimately, even after you completed an analytical test, essentially that the test performs well, with the predicted outcome, to be able to predict and verify the clinical outcome, you need a very large number of specimens.
Image Analysis
Within the context of companion diagnostic development, image analysis incorporation is felt very open-ended, but options can be validated for clinical perspective use. Image analysis can decrease time and overall cost due to the appropriate patient selection in these large datasets, which we’ve discussed previously. And it’s rapidly becoming a critical tool to support pathologists, due to the increasing complexity of biomarkers that we continue to see, particularly in immuno-oncology. As the development moves and delete or faded and more sites open up, you ensure much more and more consistency across reads and ensure that you’re getting the best data possible.
Regarding traditional pathology, it is very useful, dating back to the 16th century when the microscope was invented.
In the era of targeted oncology therapy, it can be very complicated and require a lot of information for predicting usefulness. Traditional pathology doesn’t do a good job in the diagnostic world. Digital image analysis (IA) can help. The technology is rooted in traditional pathology but there are things that the computer can do better than pathologists, like being consistent, and therefore increasing the precision of the outcome. It can simply be more accurate when it comes to counting cells or dealing quickly with a very large amount of data.
The cooperation of human pathologists and the digital domain will yield the most appropriate way to negotiate all the information that is needed for trials.
Oncologists know image analysis-based CDx is going to be designed to either be equivalent or perhaps a bit better tool for normal pathological analysis of tissue. When you’re going to send a sample across the country, potentially to be processed differently, arriving at a different laboratory, seen by a different person, and then having to be done manually, there’s going to be a lot of issues regarding reproducibility.
The computer algorithm is meant to augment or improve the ability to quantify the biomarker. It is designed to be consistent with a pathologist-trained approach, but has better precision and reproducibility over essentially, pathologists trying to come up with the same answer.
In some cases, IA may be the only adequate level of performance required to stratify patients.
It differs substantially from a manual review of a slide. Looking into the future, the complexity of targeted oncology therapies and new approaches like antibody drug conjugates (ADCs) and immuno-oncology modulators are going to drive more and more reliance on image analysis due to the complicated biology, the mechanism of action, and the complexity of the way that tissue or other samples like fluid need to be assessed.
IA for CDx is a way to improve substantially over the way we currently look at slides. From an oncologist’s point of view, when we’re thinking about staining a slide and deciding whether a patient will get lifesaving therapy or not, our answer must be as accurate and precise as possible.
It is also likely that as we continue to develop, we will eventually replace much of the manual assessment that we do today. IA is going to be the wave of the future. Just as with the retrospective analyses that are coming out of people doing more and more multiplex assays.
It is simply getting to a point where pathologists will not be able to manually score samples with such a level of complexity. The only way you will be able to reproduce and accurately analyze the huge amount of data is through IA approaches.
Leap is again a great example of that. While their assay is pretty straightforward, it is an RNAscope, a technology developed by Advanced Diagnostics, they are measuring mRNA levels. The technology is exquisitely sensitive. In theory, you can pick up one mRNA
Per sample. It can be difficult to find something that sensitive. However, a potential problem with RNAscope is that it can be very challenging for a pathologist to manually score.
You have to count the number of dots in the cells. The more dots there are, the more expression you have. This requires is the pathologist to zoom in onto the tissue to high magnification to identify if the cell has dots, then the pathologist has to zoom out, move to another section of the tissue, and continue to do that with all the zooming in and out.
It is very easy for a pathologist to lose context about the whole tissue and it is different than immunohistochemistry (IHC), which a lot of times you can just score. The pathologists scored a lower magnification and because of this, so there was concern about reproducibility. Leap needed to develop an image analysis approach that could quantify the RNAscope signal.
In some ways, the Leap assay is perfect for this, because it forms broad foci in the cells. All you need to do is develop an algorithm with software that can identify tumor cells and then count the number of foci in the cells.
Leap did some initial work on this and were able to show that with the image analysis, it was much more reproducible. If you have an image analysis algorithm that is performing well, it will perform the same way every time and should be reproducible.
By developing this image analysis with Flagship, Leap alleviated the reproducibility concern.
As part of CLIA and CAP validation, you do have to show two steps:
- Show that the wet assay is performing well
- Show that the image analysis is working well, too
For Leap Therapeutics, IA has been much more reproducible and they believe that you will see much more use of IA in the future.
Image Analysis in the Context of a Medical Device
Guidelines that are being discussed and regulatory perspective, keep in mind that once you transfer a wet assay to a digital assay solution, you essentially create a new assay. So all the traditional steps that pathologists are familiar with that go into the validation and the quality control of the assay are still true. The degree of complexity
Going from an LDT to a device, the degree of complexity of work is difficult.
When working in new territory, it is complex. Keep in mind:
- Use guiding principles of quality control when dealing with the assay
- Use a rigorous process of validating the assay
You may have to resort to the clinical outcome to measure and move through a regulatory pathway.
Within the context of clinical trials, you are often getting very limited tissue, due to the impact on the patient. But again, that’s where image analysis can help to support these large datasets.
In terms of creating the regulatory analytical framework for the assay there is a lot that goes into each phase.
- Starting with receiving the images, you need to make sure that the images have been scanned with a scanner that is compatible with the digital solution that you are creating.
- Then, going on to the development of the algorithm and verification of the vis-a-vis the manual pathologist, there is a lot to do.
When it comes to regulations for the IA, there is not much difference from regulations that apply to every assay. One thing is to ensure the analytical sensitivity and analytical specificity of the IA are acceptable and to also define what acceptable is, in the context of the intended use of the assay. You will also need to be sure that the digital assay provides enough accuracy and precision to be consistent with the clinical use of the assay
Of the six predictive IHC biomarker tests approved by the FDA, in terms of the wet assay, only one – the human epidermal growth factor receptor 2, HER2, for breast and gastric cancers – has IA quantification algorithms cleared by the FDA.
Until now, this has been considered a complex undertaking. And while it is complex, generating a digital solution not only provides you better accuracy and precision but also gives you an additional large amount of data that cannot be obtained by traditional wet IHC quantification.
For example, for PD-L1 a traditional assay may give you some information on the distribution of the PD-L1 in addition to the quantification, but a digital assay can tell you exactly like:
- How many lymphocytes are positive for the marker
- How many macrophages are positive for the marker
- What level of positivity in the macrophages is in relation to the stroma compartment
- Where are the positive lymphocytes and macrophages in relation to the tumor nest… the list could go on and on
There are regulatory gray areas and challenges in trying to embed your image analysis solution into regulatory approval, given there is no current precedent. However, because of rapid technological development, many new approaches are coming to market with a large number of targeted medicines in oncology and other therapeutic areas.
So far, we do not have image analysis alone cleared as a companion diagnostic by the FDA. But there are in-vitro diagnostic tests with image analysis quantification algorithms embedded that are cleared by the FDA.
It’s predicted that this process will just expand, and it’s all based on the rigor of the quality standards, of testing it, to use it to reproducibility and reliability, starting from analytical validation, going to an ending up with the clinical validation, and demonstrating the performance of companion diagnostic.
The contribution of image analysis to informing clinicians and making some critical medical decisions is key, and it’s all about the risk. So, that’s the background for either the traditional companion diagnostic or image analysis-based companion diagnostic, and whether this can be satisfied by pathologists or healthcare providers or not.
These gray areas will be worked out by regulatory agencies. The FDA is working actively on this. The fact that they issued guidance a year or so ago about the tools for making clinical decisions is indicative that they would like to work with sponsors in industry and academia on how to approach the best regulatory oversight, especially for image analysis.
For solutions like image analysis software, maybe only certain functions may be fitting right now, for regulatory oversight this device versus the others which are not. These are the things that we are looking to the agency for right now, and Flagship is a part of this process as well.
Get in Touch for More Information to Support Your Projects
If you are interested in a consultation with any of our experts, please contact us.
You may also view our recorded presentation about these topics. We are happy to provide a consultation on your project. If you need assistance in planning your sample and regulatory approaches and strategy, Flagship can help, too. Contact us to learn more.
Questions and Answers from the Recorded Session:
- QUESTION: Can you provide an overview of image analysis at a high level?
ANSWER: Essentially, IA replaces some pieces that are traditionally done by a pathologist. First, you have the generation of a digital image by a scanner. More and more, pathologists can use IA as a substitute for glass slides.IA also essentially analyzes the data that is contained in each slide. For this, you need cellular recognition, for which you need to create an algorithm to identify which objects on the slide are cells and which are not, then you can quantify the cell.
Next, you define cellular compartments. For instance, the tumor compartment, as defined by the aggregate of the neoplastic cells, and other cells like lymphocytes and macrophages that may be contained in that compartment. And the stroma compartment – the supporting tissue around which the tumor is organized. Then you can quantify by the computer. You can do this by enumeration, in the case you want to know many cells are positive for of the marker and not the intensity of the marker. You can also do this by quantification to measure the intensity of the marker and relate that to the protein expression in traditional IHC or RNA. Everything else is a derivative endpoint. - QUESTION: What is the difference in planning between Phase One and Phase Three from a trial perspective?
ANSWER: Phase One is much more exploratory and your assay does not need to be nearly as well validated. You want a good biomarker assay but you do not have to rigorously validate yet, because that costs a lot of money. You are still testing hypotheses and won’t have the resources to do a lot more.
Also, once you get into a Phase One clinical trial it implies that you need to have your biomarker strategy testing already in place and hopefully you have discussed it with the FDA in detail. This will give you clarity on how to start collecting your thesis samples, how you store them, and everything related. By Phase Three, your assay is being used as a test and is pretty much locked down, so that’s the one you’re banking on. At that point, you’ll much more rigorously validate and verify, spend on all GMP reagents. You’re out of your research use only mode and into investigational use only, and hopefully well on the path for a CDx.This also depends on the attributes of the investigational drug. The anticipated risk-benefit profile or anticipated risk may eventually push you toward more advancing into the assay qualifications, and validations. There are now Phase One trials that use IVDs for example, for prospective enrollment of patients, and you have to have the CLIA validation also split out in the very first portion of Phase One that you can use later on for the expansion phase. As you may know, we do not really have specific phases of clinical development. Though it begins with Phase One, if the drug is highly successful, it will move quickly. You have to be ready. You have to understand the assay, the targets, and that will determine the level of investment and development, even in Phase One in oncology.
You may also view our recorded presentation about these topics. Flagship is happy to provide a consultation on your project. If you need assistance in planning your sample and regulatory approaches and strategy, we can help, too. Contact us to learn more.